
Dartmouth College (Neuroscience Research)
Depression and anxiety disorders affect nearly one in three adults, yet traditional antidepressants often take weeks to work and fail many patients. Psilocybin, the active ingredient in 'magic mushrooms,' has emerged as a breakthrough because a single dose can lift depressive symptoms for months. For years, scientists believed these benefits came solely from the same brain 'lock' that causes hallucinations (the 2A receptor). However, in this study “The serotonin 1B receptor is required for some of the behavioral effects of psilocybin in mice,”, Fleury and Nautiyal investigate a different, overlooked 'lock' (the 1B receptor) to see if it holds the key to psilocybin's long-term healing power without the 'trip.'
Researchers compared normal mice to knockout mice, animals genetically engineered to lack the 1B receptor. They used a 'glow-in-the-dark' molecular marker to map which areas of the brain 'turned on' after psilocybin treatment. To see if the drug actually improved 'depressed' behavior, they put the mice through stress tests that mimic human anxiety and anhedonia (the inability to feel pleasure). By comparing how these two groups of mice responded, they could isolate exactly what the 1B receptor does when psilocybin enters the system.
The researchers discovered that while the 1B receptor has nothing to do with the 'hallucination' phase, it is essential for the antidepressant effects. The study revealed that psilocybin uses the 1B receptor to 'rewire' the amygdala, the brain's emotional thermostat. Without this specific receptor, psilocybin failed to reduce anxiety or restore the ability to feel pleasure. This is a first-of-its-kind discovery showing that psilocybin's ability to change a patient's mood appears to be biologically distinct from its ability to cause a psychedelic experience. Furthermore, the study highlights critical sex differences, showing these pathways are particularly influential in female biology.
This research paves the way for a new generation of 'non-trippy' antidepressants, potentially making life-changing mental health treatment accessible to millions of people who cannot, or should not undergo a traditional psychedelic experience. In recognition of the Nautiyal lab’s groundbreaking work, we have selected the Department of Psychological and Brain Sciences, Dartmouth College, as our April 16, 2026 foundation award recipient.

Harvard University Medical School (Neuroscience Research)
Alzheimer’s disease (AD) is marked by a long preclinical phase during which molecular disruptions precede symptom onset. While genetic risk factors are well studied, the contribution of environmental and nutritional factors remains less clear.
In their August 06, 2025, Nature publication, “Lithium deficiency and the onset of Alzheimer’s disease,” Aron, et al. identify endogenous lithium (Li) as a critical regulator of cognitive resilience and AD pathology. Using metallomic profiling, the team found that cortical Li levels were significantly reduced in individuals with mild cognitive impairment (MCI) and AD, even though serum Li remained unchanged. Further, Li was sequestered into amyloid-β (Aβ) plaques, depleting its bioavailability in vulnerable brain regions.
In mouse models, dietary depletion of Li (~50% reduction in cortical Li) accelerated Aβ deposition, tau phosphorylation, neuroinflammation, synaptic and myelin loss, and cognitive decline. These effects were mediated in part through hyperactivation of the kinase GSK3β. Single-nucleus RNA-seq revealed transcriptomic shifts across neurons, oligodendrocytes, astrocytes, and microglia that overlapped strongly with those observed in human AD brain tissue. Replacement therapy with lithium orotate - a Li salt that binds less avidly to Aβ - prevented or reversed pathological changes and restored memory in AD mouse models, while also preserving cognition during normal ageing.
These findings position lithium deficiency as a potentially early driver of AD pathogenesis, linking disrupted metal homeostasis to amyloid sequestration, tau pathology, neuroinflammation, and synaptic loss. Importantly, they highlight lithium orotate as a promising therapeutic avenue that avoids the toxicity associated with pharmacological Li salts while maintaining physiological brain Li levels.
In recognition of the Yankner lab’s groundbreaking work uncovering lithium deficiency as a modifiable environmental factor in the onset of Alzheimer’s disease, we have selected the Department of Genetics at Harvard Medical School as our August 30, 2025 foundation award recipient.

Harvard University (Neuroscience Research)
In their February 5, 2025, publication in Neuron, titled "Ketamine induces plasticity in a norepinephrine-astroglial circuit to promote behavioral perseverance," Duque, et al. from Harvard University and collaborating institutions investigate the long-lasting antidepressant-like effects of ketamine, particularly its ability to counter behaviors akin to "giving up." When faced with persistently futile situations, animals, like humans, can enter a passive state, analogous to learned helplessness seen in some depressive disorders. While ketamine is known for its rapid therapeutic effects, the precise brain mechanisms underlying its sustained impact on such behaviors have been a critical area of research.
Using larval zebrafish, which exhibit a "giving up" response when their swimming efforts become ineffective, the researchers demonstrated that a brief exposure to ketamine results in a long-term suppression of this passivity, promoting behavioral perseverance. Through sophisticated whole-brain imaging, they discovered a two-phase mechanism: ketamine initially triggers a strong, acute hyperactivation of a specific circuit involving norepinephrine (a key brain chemical) and astrocytes (star-shaped support cells in the brain). This astrocytic hyperactivation was found to be dependent on norepinephrine. Following this intense initial response and after ketamine was cleared from the system, the same norepinephrine-astroglial circuit showed a prolonged period of reduced responsiveness (hypoactivation) to futility cues. Optogenetic and chemogenetic manipulations confirmed that this norepinephrine-astrocyte pathway is both necessary and sufficient for ketamine's lasting effects on perseverance. Notably, the study also found that astrocytes in the mouse brain show similar activation patterns during futility-related behaviors and in response to ketamine, suggesting a conserved mechanism across species.
These findings from Duque, et al. are significant as they unveil a novel plasticity mechanism where ketamine’s transient, potent stimulation of the norepinephrine-astroglial system leads to a lasting desensitization of this circuit, ultimately promoting resilience against futility-induced passivity. This work not only deepens our understanding of how ketamine exerts its sustained antidepressant-like actions but also highlights astrocytes as crucial players in mediating these long-term behavioral changes, potentially opening new therapeutic avenues for affective disorders.
In recognition of Duque, et al.’s innovative research elucidating the dynamic interplay between ketamine, norepinephrine, and astroglial cells in promoting lasting behavioral perseverance, we have selected Harvard University as our April 16, 2025 funding recipient.

Columbia University / Taub Institute (Neuroscience Research)
In Alzheimer's disease (AD), the presence of the APOEε4 allele is one of the strongest genetic risk factors, particularly in non-Hispanic White populations. However, some individuals carrying the APOEε4 allele do not develop AD or experience a delayed onset of symptoms, suggesting the existence of protective mechanisms.
In their recent publication in Acta Neuropathologica, Bhattarai et al. from the Taub Institute for Research on Alzheimer's Disease and the Aging Brain at Columbia University identified rare genetic variations in the fibronectin 1 (FN1) gene that may protect against APOEε4-mediated AD pathology. Using whole-genome sequencing data from multiple cohorts, the researchers found that two missense variants in FN1 were present in cognitively unaffected APOEε4 homozygous carriers but absent in affected carriers with clinically diagnosed AD.
The study provides compelling evidence that FN1 deposition increases with APOEε4 dosage and that unaffected homozygous APOEε4 carriers have FN1 deposition levels similar to non-carrier controls. Furthermore, using a zebrafish model, they demonstrated that knockout of the FN1 ortholog alleviates amyloid toxicity-related pathological changes.
These findings suggest that basement membrane thickening and remodeled extracellular matrix composition in the blood-brain barrier may contribute to APOEε4-mediated AD pathology. Importantly, this research opens new avenues for potential therapeutic interventions targeting the blood-brain barrier basement membrane to mitigate the impact of APOEε4-mediated risk of AD.
In recognition of the Vardarajan, Kizil, and Mayeux labs' innovative cross-species approach to uncover protective mechanisms against one of the strongest genetic risk factors for AD, we have selected the Taub Institute for Research on Alzheimer's Disease and the Aging Brain at Columbia University as our August 30th, 2024 funding recipient.

Washington University in St. Louis
In their recent publication, Long non-coding RNA SNHG8 drives stress granucle formation in tauopathies, in Molecular Psychiatry, the Karch Lab at Washington University in St. Louis has made significant strides in understanding the molecular mechanisms underlying tauopathies, a class of neurodegenerative diseases characterized by the accumulation of hyperphosphorylated tau protein.
In this study, Bhagat et al. utilized induced pluripotent stem cell (iPSC)-derived neurons carrying mutations in the MAPT gene, which encodes the tau protein, to investigate the role of long non-coding RNAs (lncRNAs) in tauopathy pathogenesis. Through a comprehensive transcriptomic analysis, they identified a set of 15 lncRNAs that were commonly dysregulated across three different MAPT mutations.
Among these lncRNAs, the researchers focused on SNHG8, which was found to be significantly downregulated in a mouse model of tauopathy and in brain samples from patients with frontotemporal lobar degeneration, progressive supranuclear palsy, and Alzheimer's disease. Importantly, they demonstrated that SNHG8 interacts with tau and stress granule-associated RNA-binding proteins, such as TIA1, FUS, DDX3X, and TDP-43.
Remarkably, Bhagat et al. revealed that overexpression of mutant tau leads to reduced SNHG8 expression and induces stress granule formation, a pathological hallmark of tauopathies. Conversely, rescuing SNHG8 expression resulted in reduced stress granule formation and decreased levels of TIA1, a key regulator of stress granule assembly.
This work by the Karch Lab has uncovered a novel mechanism by which SNHG8 dysregulation contributes to the formation of stress granules in tauopathies, shedding light on the intricate interplay between lncRNAs, tau, and RNA-binding proteins in the pathogenesis of these devastating diseases.
In recognition of the Karch Lab for their exceptional research, we have selected Washington University in St. Louis as our April 16th, 2024 funding recipient.
Washington University in St Louis (Neuroscience Research)
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia in older adults. One of the biggest challenges in treating AD is that neurodegeneration starts years before symptoms appear. Identifying biomarkers that can detect preclinical AD would allow for early intervention, which is presumed to be more effective than treatment later in disease progression.
In their recent publication in Science Translational Medicine, Ferreiro et al. report a potential microbiome biomarker signature that distinguishes individuals with preclinical AD from healthy controls. Using multi-omic analysis, the researchers identified specific gut bacteria and microbial metabolites that were altered in older adults who showed AD pathologies on PET scans but did not yet show cognitive impairment. If validated in larger cohorts, these microbial metabolites could enable non-invasive screening to identify high-risk individuals in early, pre-symptomatic stages when disease-modifying therapies are more likely to be effective.
In recognition of the Dantas lab's use of multi-omics to uncover the gut-brain axis perturbations that precede dementia, we have selected Washington University in St. Louis as our August 30th, 2023 funding recipient.

Northwestern Feinberg School of Medicine
In a subset of major depressive disorder (MDD) patients, chronic dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis occurs, resulting in increased levels of the ‘stress hormone’ cortisol during the normal rest period (i.e., evening and night). However, the mechanistic relationship between chronically elevated resting cortisol and behavioral deficits in motivation and reward processing remains unclear. Given that women are diagnosed with MDD at twice the rate of men, it is important to understand whether the mechanisms linking cortisol to the symptoms of MDD differ by sex. In their February 2023 publication Holloway et al. in Neuropsychopharmacology used subcutaneous implants in mice to chronically elevate free plasma corticosterone (the rodent homolog of cortisol; 'CORT') during the rest period in males and females and examined changes in behavior and dopamine system function.
The researchers concluded that chronic CORT dysregulation impairs motivation by impairing dopaminergic transmission in the DMS, but via different mechanisms in male and female mice, and suggest that better understanding of these sex-specific mechanisms could lead to new directions in MDD diagnosis and treatment.
In recognition of the Lerner lab's compelling evidence that males and females can display different underlying mechanisms to achieve similar functional or behavioral outcomes and their suggestion that sex differences at the molecular level must be considered to appropriately translate preclinical discoveries into medicines, we have selected Northwestern Feinberg School of Medicine as our April 16th, 2023 funding recipient.

Salk Institute for Biological Studies (Neuroscience Research)
In 2016, Kay Tye and colleagues at the Salk Institute for Biological Studies discovered that a group of neurons in the brain’s basolateral amygdala (BLA) helps assign valence when mice are learning. One set of BLA neurons was activated with positive valence, as the animals learned to associate a tone with a sweet taste. A separate set of BLA neurons was activated with negative valence, as the animals learned to associate a different tone with a bitter taste.
Their recent research, published in Nature on July 20, 2022, went deeper into the mechanism of associating positive or negative valence through new learning by using CRISPR gene editing approaches to selectively remove the gene for neurotensin from the cells. Without neurotensin signaling in the BLA, mice could no longer assign positive valence and didn’t learn to associate the first tone with a positive stimulus. While the absence of neurotensin did not block negative valence. The animals instead became even better at negative valence, having a stronger association between the second tone and a negative stimulus. The findings suggest that the brain’s default state is to have a bias toward fear—the neurons associated with negative valence are activated until neurotensin is released, switching on the neurons associated with positive valence. High levels of neurotensin promoted reward learning and dampened negative valence.
In recognition of the Tye lab's ability to manipulate this switch to turn on positive or negative learning (and thereby potentially impact anxiety and PTSD), we have selected the Salk Institute for Biological Studies as our August 30th, 2022 funding recipient.

Weill Institute for Neuroscience at University of California, San Francisco
Deep brain stimulation is a promising treatment for neuropsychiatric conditions such as major depression. In their October 2021 publication Scangos et al. in Nature Medicine describe a method to optimize the therapy by using multi-day intracranial electrophysiology and focal electrical stimulation to identify a personalized symptom-specific biomarker and a treatment location where stimulation improved symptoms. They then implanted a chronic deep brain sensing and stimulation device and implemented a biomarker-driven closed-loop therapy in an individual with treatment-resistant depression, resulting in a rapid and sustained improvement.
In recognition of the Krystal & Stark labs' innovative methods to optimize DBS through personalization, we have selected the Weill Institute for Neuroscience at the University of California, San Francisco as our April 16th, 2022 funding recipient.

Center for the Neurobiology of Learning and Memory at University of California, Irvine
The overexpression of calcineurin leads to astrocyte hyperactivation, neuronal death, and inflammation, which are characteristics often associated with pathologic aging and Alzheimer’s disease. Using a canine model, Radhakrishnan, et al. at the University of California, Irvine have shown that tacrolimus, a calcineurin inhibitor commonly used to suppress transplant-organ rejection, prevents age-associated microstructural atrophy, which they measured using higher-order diffusion MRI, in the middle-aged beagle brain; supporting the idea that calcineurin inhibitors may have the potential to prevent aging-related pathology if administered at middle age.
In recognition of the Stark & Head labs' identification - published 09-June-2021 in The Journal of Neuroscience - of a possible intervention (using an already FDA approved drug) to delay or minimize age-related neurodegeneration and dementia, we have selected the Center for the Neurobiology of Learning and Memory at University of California, Irvine as our August 30th, 2021 funding recipient.

Medical College of Georgia at Augusta University
Researchers at the Medical College of Georgia at Augusta University have shown that chronic unpredictable stress, an animal model of depression, decreases spontaneous firing rates, increases firing irregularity and alters the firing properties of AgRP neurons in the arcuate nucleus (ARC) in both male and female mice. Fang, et al. found that chemogenetic inhibition of AgRP neurons increases susceptibility to subthreshold unpredictable stress, and that conversely, chemogenetic activation of AgRP neurons completely reverses anhedonic and despair behaviors induced by chronic unpredictable stress.
The 11-Jan-2021 Molecular Psychiatry publication, suggests that AgRP neurons in the ARC are a key component of neural circuitry involved in mediating depression-related behaviors and that increasing AgRP neuronal activity could be a novel and effective treatment for depression.
In recognition of the Lu lab's identification of cellular processes and potential therapeutic target related to depression, we have selected the Medical College of Georgia at Augusta University as our April 16th, 2021 funding recipient.

Michigan State University (Neuroscience Program)
It is hypothesized that factors promoting resilience to stress may offer treatment strategies for disorders such as anxiety and depression. Using a physical-restraint mouse model, Yang, et al. found that a neurospecific synaptic enzyme, type 1 adenylyl cyclase (Adcy1), that positively regulates the cAMP signaling cascade is related to molecular stability and behavioral resilience. The researchers demonstrated that transgenic overexpression of Adcy1 not only rescued behavioral resilience in Adcy1-tg mouse, but also prevented the physical restraint-induced down-regulation of brain-derived neurotrophic factor (BDNF) and neuropeptide Y (NPY).
In recognition of the Wang lab's demonstration of a novel function of Adcy1 in stress coping, suggesting Adcy1 as a potential target to antagonize stress vulnerability and promote antidepressant efficacy, we have selected Michigan State University as our August 30th 2020 funding recipient.

University of California San Diego (Neuroscience Research)
In their January 2020 publication, Prakash, et al. explore the molecular mechanisms underlying susceptibility and resilience to stress-induced anhedonia. Their experiments in a rat model showed that those susceptible, but not resilient, displayed an increased number of neurons expressing the biosynthetic enzyme for serotonin, tryptophan-hydroxylase-2 (TPH2), in the ventral subnucleus of the dorsal raphe nucleus (DRv) ultimately revealing that activation of amygdalar CRH+ neurons induces resilience, and suppresses the gain of serotonergic phenotype in the DRv that is characteristic of susceptible rats.
In recognition of the Dulcis Lab's identification of a novel molecular marker of susceptibility to stress-induced anhedonia (a core symptom of depression) and a means to modulate it, we have selected the University of California San Diego as our April 16th 2020 funding recipient.

Yale University School of Medicine (Neuroscience Research)
Rapid-acting antidepressants, such as ketamine, have been previously shown to require stimulation of mTORC1 signaling - which is regulated by multiple signals, including sestrin.
In their June 2019 publication, Kato, et al. suggest that NV-5138, a highly selective small molecule modulator of sestrin that penetrates the blood-brain barrier, produces rapid and long-lasting antidepressant effects and rapidly reverses anhedonia caused by chronic stress exposure.
In recognition of the Duman Lab's investigation of the mechanism of NV-5138 in producing rapid synaptic and antidepressant behavioral responses, we have selected the Yale University School of Medicine as our August 30th, 2019 funding recipient.
Columbia University / Taub Institute (Neuroscience Research)
The accumulation of tau proteins during the early stages of Alzheimers Disease impairs and eventually kills neurons. The susceptibility of neurons to tau accumulation is greater in excitatory neurons than in inhibitory neurons and is related to the cells ability to clear excess tau.
In their Dec 2018 publication, Fu, et al. suggest that BCL2-associated athanogene 3 (BAG3), is a hub, or master regulator, gene of autophagy. Their research verified that reducing BAG3 levels in primary neurons exacerbated pathological tau accumulation, whereas BAG3 overexpression attenuated it. Their work thus defined a tau homeostasis signature that underlies the cellular and regional vulnerability of excitatory neurons to tau pathology.
In recognition of the Duff Labs important insights into the mechanism of tau homeostasis in the neurodegenerative brain, we have selected the Taub Institute for Research on Alzheimers Disease and the Aging Brain as our April 16th, 2019 funding recipient.

Drexel University (Neuroscience Research)
Because histone acetylation homeostasis is critical for mediating epigenetic gene control throughout neuronal development, its misregulation may contribute to cognitive impairment preceding Alzheimers Disease (AD) pathology.
In their May 2018 publication, Panikker, et al. suggest that disruption of the Tip60 HAT/HDAC2 balance is a critical initial step in AD. They report that this disruption of Tip60 HAT/HDAC2 homeostasis occurs early in the AD Drosophila brain and triggers epigenetic repression of neuroplasticity genes before A-beta plaques form. Increasing Tip60 in the AD brain restores Tip60 HAT/HDAC2 balance, reverses neuroepigenetic alterations to activate synaptic genes, and reinstates brain morphology and cognition.
In recognition of the Elefant Lab's important insights into the mechanism of epigenetic transcriptional repression in the neurodegenerative brain, we have selected Drexel University (Neuroscience Research) as our August 30th, 2018 funding recipient.

University of Texas Southwestern Medical Center (Neuroscience Research)
Studies suggest that dendritic spine loss, a characteristic of early Alzheimers disease (AD), is induced by soluble, multimeric amyloid-Beta 42 (AB-42), which, through postsynaptic signaling, activates the protein phosphatase calcineurin. An earlier retrospective study showed that human transplant patients receiving FK506, an inhibitor of calcineurin, developed AD at a much lower incidence than expected. But since a calcineurin inhibitor such as FK506 can produce serious immunosuppressant and carcinogenic side effects, a more complete understanding of molecular mechanisms will be necessary to identify more specific targets for more precisely targeted drugs.
In their March 2018 publication, Stallings, et al. report that Pin1 is a critical downstream dephosphorylation target of AB-42-calcineurin signaling. The researchers report that knockout of Pin1 or exposure to AB-42 induced the loss of mature dendritic spines, but that that spine loss could be prevented by the addition of exogenous Pin1. Interestingly the calcineurin inhibitor FK506 blocked dendritic spine loss in AB-42-treated wild-type cells but had no effect on Pin1-null neurons. These data implicate Pin1 in dendritic spine maintenance and synaptic loss in early Alzheimer’s disease.
In recognition of the Malter Lab's important insights into the mechanism of calcineurin inhibitors in resistance to AD-related neurodegeneration, we have selected University of Texas Southwestern Medical Center (Neuroscience Research) as our April 16th, 2018 funding recipient.

Weill Cornell Medicine (Neuroscience Research)
The CACNA1C gene, encoding the Cav1.2 subunit of L-type calcium channels, has emerged as a candidate susceptibility gene for multiple neuropsychiatric disorders including bipolar disorder, schizophrenia, major depressive disorder, and autism spectrum disorder. Within the CACNA1C gene, disease-associated single-nucleotide polymorphisms have been associated with impaired social and cognitive processing and altered prefrontal cortical structure and activity.
In their August 2017 publication, Kabir, et al. report that knock-out mice harboring loss of cacna1c in excitatory glutamatergic neurons of the forebrain (fbKO) and adult mice with focal knockdown of cacna1c exhibit anxiety-like behavior and display a social behavioral deficit. But systemic treatment with ISRIB, a small molecule inhibitor that suppresses the effects of phosphorylated eIF2-alpha on mRNA translation, was sufficient to reverse the social deficit and elevated anxiety-like behavior in the adult cacna1c fbKO mice.
In recognition of the Rajadhyaksha lab's identification of a novel mechanism and potential future therapeutic target for anxiety and other neuropsychiatric-related behaviors we have selected Weill Cornell Medicine (Neuroscience Research) as our August 30, 2017 funding recipient.
Washington University in St Louis (Neuroscience Research)
Because the accumulation of hyperphosphorylated tau directly correlates with cognitive decline in Alzheimer's disease, a promising therapeutic strategy may be to reduce total tau expression. Researchers from the Miller Lab at Washington University in St Louis have identified anti-sense oligonucleotides (ASOs) that selectively decrease human tau mRNA and protein in mice engineered to express mutant P301S human tau.
In their January 2017 publication, DeVos, et al. report that "After reduction of human tau in this mouse model of tauopathy, fewer tau inclusions developed, and preexisting phosphorylated tau and Thioflavin S pathology were reversed. The resolution of tau pathology was accompanied by the prevention of hippocampal volume loss, neuronal death, and nesting deficits. In addition, mouse survival was extended, and pathological tau seeding was reversed."
While showing beneficial effects at the molecular, cellular, anatomical, and behavioral levels in the mouse model, DeVos, et al. also showed that their tau-targeted ASOs reduced tau mRNA and protein in the brain, spinal cord, and cerebrospinal fluid of non-human primates (cynomolgus monkeys).
In recognition of these important insights into the therapeutic possibilities of tau reduction in both mouse and primate models we have selected Washington University in St Louis School of Medicine (Neuroscience Research) as our April 16th, 2017 funding recipient.
Salk Institute for Biological Studies (Neuroscience Research)
Although beta-amyloid (A-beta) has been associated with Alzheimers Disease since the initial characterization of the disease, the mechanistic relationship between intracellular amyloid, aging, and neurodegeneration is not well understood. Using a human central nervous system cell line that conditionally expresses A-beta, researchers from the Schubert lab at the Salk Institute have characterized a distinct form of nerve cell death caused by intracellular A-beta in which A-beta induces the expression of multiple proinflammatory genes and an increase in both arachidonic acid and eicosanoids.
Through identifying the molecular basis of this inflammatory response Currais, et al. (23-June-2016) demonstrate that intracellular A-beta accumulation and this early form of proteotoxicity can be blocked by the activation of cannabinoid receptors, with tetrahydrocannabinol showing a particularly potent protective effect.
In recognition of these important insights into the inflammatory pathways associated with Alzheimers Disease pathology and possible therapeutic approaches we have selected The Salk Institute for Biological Studies (Neuroscience Research) as our August 30th, 2016 funding recipient.

Johns Hopkins University School of Medicine (Neuroscience Research)
A major limitation of current antidepressant drug therapy is the lag of weeks to months before attaining any therapeutic benefit. Moreover, a significant percentage of depression patients experience no benefit from these drugs after any period of time.
Ketamine has been shown to relieve clinical depressive symptoms in as little as 2 hours with therapeutic effects lasting for a period of weeks, even in otherwise drug resistant cases of depression. However ketamine, an anesthetic, can produce serious side effects including hallucinations, dissociative dream-like state, and possibly addiction. Producing a new generation of antidepressant drugs that deliver ketamine's rapid and long-acting effects while minimizing side effects and potential for abuse will require a detailed understanding of its molecular mechanism of action.
In March 2016 Harraz, et al. of Johns Hopkins University published a report in which they used biochemical, neuronal cell culture, and mouse behavioral studies to describe a novel signaling pathway in which ketamine stabilizes a small G protein, Rheb, which activates mTOR to exert antidepressant actions. In recognition of their efforts to begin to describe the molecular mechanisms involved in the rapid and lasting antidepressant effects of ketamine, we have selected Johns Hopkins University School of Medicine (Neuroscience Research) as our April 16th, 2016 funding recipient.

University of Kentucky College of Medicine (Neuroscience Research)
(Neuroscience Research)Researchers at the University of Kentucky published results last month describing a connection between memory impairment and declining levels of a naturally occurring protein involved in immune response and calcium regulation in neurons, called FK506 Binding Protein. This UK study follows a June 2015 University of Texas Medical Branch publication showing, for the first time in humans, almost complete protective effects against the development of dementia in organ transplant recipients who have been receiving calcium-regulating immunosuppressant drugs such as FK506. In support of their continued efforts to discover the molecular mechanisms involved in neuronal calcium regulation and its connection to age and disease-related memory loss, we have selected the University of Kentucky College of Medicine (Neuroscience Research) as our August 30th 2015 funding recipient.
Animal models in neuroscience research have long been indispensable in deciphering cellular mechanisms of the Central Nervous System and how those mechanisms are altered by disease. In recent years studies on mice and rats (treated or genetically engineered to develop the same kinds of cellular, cognitive, and behavioral problems as seen in Alzheimerís Disease) have produced a model which ties the neurodegeneration and cognitive decline associated with Alzheimerís Disease (AD) to an overabundance of intracellular calcium. In this model, this imbalance in neuronal calcium induces multiple cellular changes including the hyperactivation of an abundant cellular protein called Calcineurin which, in turn, activates other proteins and sets in motion a cascade of reactions that ultimately account for the synapse loss, memory impairment, and other symptoms associated with dementia.
With Calcineurin playing a central role in the calcium-focused model, researchers have found that treating AD mice with an inhibitor of Calcineurin's ability to activate other proteins slowed, prevented, and even reversed synapse loss and memory impairment, lending support to the model. These reports, as interesting and encouraging as they may be, represent only a few of the many times that Alzheimerís disease has seemingly been cured in rodents. Transferring these successes to human patients, however, has yielded little clinical success.
But in June 2015 Luca Cicalese and colleagues at the University of Texas Medical Branch (UTMB) published data from humans that appears to give strong support to the proposed Calcineurin-dependent animal model. It turns out that Calcineurin-inhibitors such as FK506 and Cyclosporin-A have been used for decades in organ transplant recipients as anti-rejection immunosuppressant drugs. The UTMB group compared the prevalence of clinically diagnosed dementia in 2644 transplant patients who have been taking Calcineurin-inhibitors with that seen in the general public. While prevalence numbers in the youngest group (<65 years old) were both very low, the impact of the Calcineurin-inhibitor treatment was dramatically apparent in older groups.
For people <65, dementia is diagnosed in 0.07% of the general population and 0.09% of the Calcineurin-inhibitor treated group(n=2/2057).
-
For people >65, dementia is diagnosed in 11% of the general population but only 1.02% of the Calcineurin-inhibitor treated group (n=6/587).
-
For people >75, dementia is diagnosed in 15.3% of the general population but only 0.6% of the Calcineurin-inhibitor treated group (n=1/149).
-
And in the >85 age cohort, dementia is diagnosed in 32% of the general population but was undetected in the Calcineurin-inhibitor treated group (n=0/14).
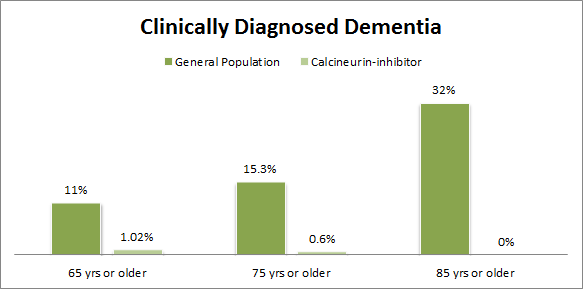
That actual human disease prevalence data gives such strong support to the animal-based disease model marks an important milestone in Alzheimerís disease research, but the work is far from finished. The Calcineurin-inhibitor immunosuppressant drugs, such as FK506, that seemingly so effectively prevent the development of dementia in humans have serious side effects, including increased risk of infection and malignancies. For thousands of organ transplant recipients these risks are justified, but they would not be so for the general population. Rather, the calcium regulation model will need to be better understood so that more targeted drugs or other therapies that prevent neurodegeneration and memory loss while avoiding the immunosuppressive and carcinogenic side effects can be identified.
John Gant and colleagues at the University of Kentucky (UK) have provided a powerful example of further exploring the model with their 29-July-2015 publication describing a reversal of calcium imbalance and memory impairment in aging rats by overespressing (through genetic engineering) FKBP1b (FK506 Binding Protein), a protein that naturally declines with age and in Alzheimer's Disease patients. FKBP1b stabilizes intracellular calcium levels and binds to and mediates the activity of the Calcineurin-inhibitor FK506, one of the immunosupressant drugs in the UTMB study. The UK study's Principal Investigator, Philip Landfield, described the work as showing "...FKBP1b is a master regulator of calcium in brain cells, and when we restore it, it restores the regulation of calcium and dramatically improves learning in the aged animals."
To encourage such continued research in revealing the key molecules and pathways involved in the memory impairment and neurodegeneration observed in Alzheimer's Disease, we have chosen to support neuroscience research at the University of Kentucky College of Medicine through our 30-August-2015 research funding.

University of Louisville Depression Center
-targeting their projects in Cortical Mechanisms of Depression and Cognitive Neuroscience Research in Mood Disorders

UCI - MIND (Alzheimers Disease Excellence Fund)
-funding pilot research projects for junior investigators to develop preliminary data in the preparation of a formal NIH grant application.
- About McAlpine Foundation Contact © 2026